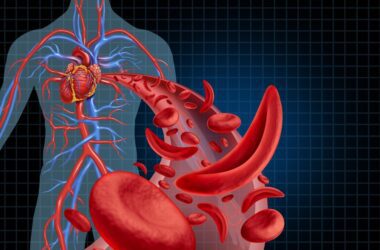





Millions of individuals worldwide are impacted by the inherited blood condition known as sickle cell anaemia (SCA). The abnormal sickle-shaped red blood cells that are produced are its defining feature. These cells are less flexible and have the potential to obstruct blood flow, leading to discomfort, exhaustion, and a host of other health issues. Having SCA makes daily life difficult and requires strength, resiliency, and access to quality care.
Epidemiology
SCA is an autosomal recessive disorder, meaning both parents must carry the sickle cell trait for a child to be born with the disease. The sickle cell trait itself is relatively common, affecting approximately 8-10% of the population in sub-Saharan Africa and 1-2% of African Americans.The global prevalence of SCA is estimated to be approximately 7.74 million people. Countries bordering the Mediterranean Sea have a significant population with SCA, with estimates ranging from 3-15%.Countries with African ancestry have a higher prevalence of SCA, with estimates ranging from 2-10%.Although less prevalent, SCA significantly impacts communities of African descent in these regions.
Etiology
Haemoglobin in sickle cell disease patients is distinct. Haemoglobin S (Hb S) is present in sickle cell disease patients with the most prevalent variation. The accepted designation for normal hemoglobin is Hb A. Compared to regular hemoglobin, hemoglobin S contains valine as the sixth amino acid in the β-polypeptide, instead of glutamic acid. Individuals who have homozygous hemoglobin S experience a variety of issues, such as anemia.
A person with sickle cell trait is heterozygous for the sickle cell gene, meaning they carry the gene. Most of the time, these persons have no symptoms. Sickle cell trait is more prevalent in ethnic groups that originated in malaria-endemic regions and offers some protection against the disease. There is a one in four probability that the child of a father with the trait and a mother with the trait will develop sickle cell disease.
Pathophysiology
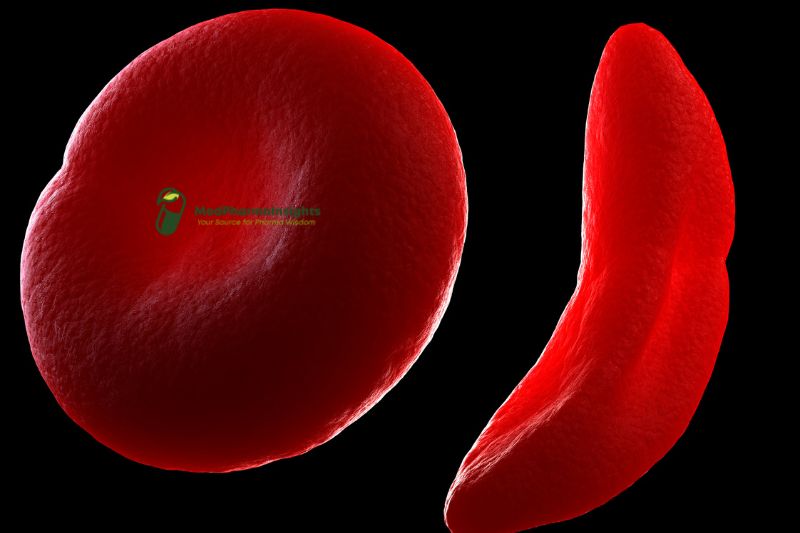
Dehydration occurs inside the cells when the membrane of red blood cells carrying hemoglobin S is weakened. Furthermore, polymerization of hemoglobin S takes place in the patient’s blood upon deoxygenation, resulting in the formation of a semisolid gel. These two mechanisms combine to generate sickle cells, which are shaped like a crescent. Compared to regular cells, sickle cells are less flexible; normal cells can move through the microcirculation because of their flexibility. Because of the rigidity, there is less blood flow via the microcirculation, which causes local tissue hypoxia.
An accelerated rate of red cell lysis in the spleen (hemolysis) causes anemia. Patients with sickle cell disease may have some fetal hemoglobin (Hb F) in their red blood cells. These don’t develop into sickle cells.
Sign and Symptoms
General Symptoms:
- Anemia: This is the most common symptom, caused by the lack of healthy red blood cells. Signs include extreme tiredness (fatigue), shortness of breath, dizziness, and irregular heartbeat.
- Swelling of hands and feet: This occurs due to blocked blood flow in these areas.
- Jaundice: Yellowing of the skin, eyes, and mouth happens when the body breaks down too many red blood cells.
Pain Crisis:
- This is a major symptom, characterized by episodes of severe pain in the bones, muscles, and joints. Pain crises can last for days or even weeks.
- Triggers include infections, cold weather, dehydration, and stress.
Other Symptoms:
- Acute chest syndrome: This serious complication involves inflammation of the lungs and can cause chest pain, cough, and difficulty breathing.
- Splenic sequestration: This occurs when the spleen traps red blood cells, leading to sudden anemia and pain in the upper left abdomen.
- Stroke: Sickle cells can block blood flow to the brain, leading to stroke.
- Delayed growth and puberty: Due to chronic anemia, children with sickle cell anemia may experience stunted growth and delayed puberty.
- Eye problems: Damage to the retina can occur, leading to vision problems.
- Increased risk of infections: Individuals with sickle cell anemia are more prone to infections due to a weakened immune system.
- Gallstones: The breakdown of red blood cells can lead to gallstones, causing pain in the upper right abdomen.
Diagnostic Tests for Sickle Cell Anemia
Several diagnostic tests are available to detect and confirm sickle cell anemia (SCA). These tests can be categorized into prenatal and postnatal tests.
Prenatal Tests:
- Chorionic villus sampling (CVS): This test involves collecting a small sample of tissue from the placenta for genetic testing. It can be performed between 11 and 14 weeks of pregnancy.
- Amniocentesis: This test involves collecting a sample of amniotic fluid, the liquid surrounding the fetus, for genetic testing. It is typically performed after 15 weeks of pregnancy.
Postnatal Tests:
- Hemoglobin electrophoresis: This is the most common and reliable test for diagnosing SCA. It separates the different types of hemoglobin in the blood, allowing for identification of abnormal sickle hemoglobin.
- High-performance liquid chromatography (HPLC): This test is similar to electrophoresis but provides more precise results.
- Sickle cell solubility test: This test measures the amount of hemoglobin that precipitates (becomes insoluble) under low oxygen conditions. This can be helpful in differentiating between SCA and other hemoglobin disorders.
- DNA testing: This is the most definitive test for diagnosing SCA and can be used to identify carriers of the sickle cell trait.
Other Tests:
- Complete blood count (CBC): This test measures the number and types of blood cells. It can be used to identify anemia and other abnormalities associated with SCA.
- Reticulocyte count: This test measures the number of immature red blood cells. It can be used to monitor the severity of SCA and to assess the response to treatment.
- Blood oxygen levels: This test measures the amount of oxygen in the blood. It can be used to diagnose and monitor complications of SCA, such as acute chest syndrome.
Treatment:
Pneumococcal infections are common in sickle cell disease patients. Prophylactic antibiotics have been demonstrated to be beneficial in a number of investigations. Adults who are allergic to penicillin are typically prescribed erythromycin along with Penicillin V 250 mg twice daily. Pneumococcal and Haemophilus influenzae vaccinations are now widely administered. Because red cell turnover is so great, folic acid is frequently used.
There have been attempts to lower the amount of hemoglobin S and raise the amount of hemoglobin F in the blood. Various medications (Box 49.9) have been demonstrated to increase fetal hemoglobin synthesis, albeit in animal models only in some cases.
Despite its potential to decrease the frequency of crises, hydroxycarbamide’s effectiveness is restricted due to its cytotoxicity.
Although it hasn’t been thoroughly proven in humans, erythropoietin has been shown in some animal models to raise hemoglobin F. The percentage of hemoglobin S has also been reduced through transfusions and exchange transfusions. This is restricted by the typical side effects of long-term infusions: sensitization, iron overload, and the possibility of blood-borne viral transmission.